| Issue |
A&A
Volume 704, December 2025
|
|
|---|---|---|
| Article Number | A73 | |
| Number of page(s) | 9 | |
| Section | Atomic, molecular, and nuclear data | |
| DOI | https://doi.org/10.1051/0004-6361/202557216 | |
| Published online | 03 December 2025 | |
Exploring chemical pathways for the interstellar molecule HOCS+: Preferential formation of the O-protonated carbonyl sulfide isomer
1
Computational Chemistry Group, Departamento de Química Física y Química Inorgánica, Facultad de Ciencias, Universidad de Valladolid,
47011
Valladolid,
Spain
2
Centro de Astrobiología (CAB), CSIC-INTA,
Carretera de Ajalvir km 4, Torrejón de Ardoz,
28850
Madrid,
Spain
★ Corresponding author: This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
12
September
2025
Accepted:
24
October
2025
Abstract
Context. The recent interstellar detection of the high-energy O-protonated carbonyl sulfide isomer (HOCS+) toward the molecular cloud G+0.693-0.027 contrasts with the non-detection of its lower-energy S-protonated counterpart, HSCO+, the global minimum in energy. This raises questions regarding the occurrence of selective formation pathways of these [H,C,S,O]+ isomers in space.
Aims. In this work, we aim to explore the most likely gas-phase formation routes for both HOCS+ and HSCO+ beyond the direct protonation of OCS (i.e., HCS+ + OH, HCO+ + SH, HOC+ + SH, and HCO + SH+) to help rationalize previous observational results. Methods. We first explored the thermodynamic feasibility of the aforementioned reactions using high-level double-hybrid B2PLYPD3∕aug-cc-pVTZ and CCSD(T)-F12∕cc-pVTZ-F12 computations. For the reaction HCS+ + OH, found to be the most thermodynamically favorable, we characterized the stationary points on its corresponding potential energy surface (PES). In addition, we also used a composite approach to refine relative energies and employed the statistical rate theory and master equation simulations to estimate rate constants and branching ratios.
Results. We show that HOCS+ is preferentially formed through the reaction of HCS+ with OH, providing a plausible chemical explanation for its interstellar presence and the non-detection of the low energy isomer. Nevertheless, while the branching ratio computed at a T ~Tkin(G+0.693) = 70-140 K is qualitatively consistent with the observations, its value is two orders of magnitude larger than the derived HOCS+/HSCO+ lower limit observational ratio (of ≥2.3). This suggests that if the upper limit of HSCO+ is close to the real abundance, additional formation pathways may also play a significant role in shaping the isomeric ratio.
Conclusions. These results highlight that including all isomers in a given family, along with their isomer-preferential formation pathways, in astrochemical models, which are in many cases isomer-insensitive, is essential to understand their formation routes.
Key words: astrochemistry / molecular data / ISM: molecules / ISM: individual objects: G+0.693-0.027
© The Authors 2025
 Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article is published in open access under the Subscribe to Open model. This email address is being protected from spambots. You need JavaScript enabled to view it. to support open access publication.
1 Introduction
Deciphering the molecular inventory of the interstellar medium (ISM) and elucidating how it is built up remains a central theme in modern astrochemistry. Sulfur-bearing molecules, in particular, provide valuable insights due to their role in stellar nucleosynthesis and the chemical evolution of galaxies (Perdigon et al. 2021), their involvement in atmospheric processes (e.g., Krasnopolsky 2012; Gómez Martín et al. 2017; Chang et al. 2023), and their potential links to early biochemical pathways, placing them at the crossroads of astrochemical and astrobiological research (Richardson et al. 2013; Todd 2022).
One of the most recently detected interstellar S-bearing molecules is oxygen-protonated carbonyl sulfide, HOCS+ (Sanz-Novo et al. 2024a). This cation was discovered toward G+0.693-0.027 (hereafter G+0.693), a shock-driven Galactic center molecular cloud that hosts a remarkably rich inventory of S-bearing species (Rodríguez-Almeida et al. 2021; Rey-Montejo et al. 2024; Sanz-Novo et al. 2024a,b; Sanz-Novo et al. 2025), using an ultra-deep molecular line survey collected with the Yebes 40m and IRAM 30m radio telescopes (Rivilla et al. 2023). The detection of HOCS+ was based on the observation of multiple a-type Ka = 0 transitions, most of which were first identified in the astronomical data and later confirmed through new laboratory spectroscopic measurements (Lattanzi et al. 2024). Interestingly, the detection of molecular ions such as HOCS+ and PO+ provides valuable indirect constraints on the cosmic-ray (CR) ionization rate affecting the cloud, as inferred through chemical modeling. Specifically, in G+0.693, enhanced CR ionization rates (i.e., 10−14−10−15 s−1; several orders of magnitude higher than the canonical Galactic disk value of ζ = 1.3×10−17 s−1) are required to reproduce their observed abundances using gas-grain astrochemical models (Rivilla et al. 2022; Sanz-Novo et al. 2024a).
The S-protonated ion, HSCO+, the global minimum in energy within the [H,C,S,O]+ isomeric family, remains unidentified in the ISM despite being more favored thermodynamically than the O-protonated form (i.e., 4.9 kcal mol−1 or 2466 K lower in energy than HOCS+; Wheeler et al. 2006; McCarthy & Thaddeus 2007; Lattanzi et al. 2018)1. Sanz-Novo et al. (2024a) derived an HOCS+/HSCO+ lower limit ratio of ≥2.3, which suggests that the formation of HOCS+ is significantly more efficient under ISM conditions.
In principle, both isomers may form via ion-molecule reactions that start with the ubiquitous carbonyl sulfide (OCS; Goldsmith & Linke 1981; Li et al. 2015; Boogert et al. 2015). Indeed, the models presented by Sanz-Novo et al. (2024a) showed that one of the dominant chemical routes is the direct protonation of OCS, involving protons from diverse sources (e.g., H3+, HCO+, and also CH3+ and CH+), in agreement with previous studies (Turner et al. 1990; Liu et al. 2002; Vidal et al. 2017; Tinacci et al. 2021). The reaction rate coefficients for these routes are available in the literature and fall within the typical range for fast protonation reactions of neutral molecules, i.e., 1.9 × 10−9 and 1.1 × 10−9 cm−3 s−1 for the first two reactions (Rakshit 1982; Adams et al. 1978), and slightly slower values of 0.8 × 10−10 cm−3 s−1 and 8.5 × 10−10 cm−3 s−1 for the later two (Smith & Adams 1978). However, we note that these studies do not distinguish between the O- and S-protonated isomers, as the measurements are based on mass spectrometry.
Alternatively, as highlighted by Sanz-Novo et al. (2024a), if we consider formation pathways analogous to those proposed for the isovalent HOCO+ (see, e.g., Fontani et al. 2018; Majumdar et al. 2018; Harada et al. 2022), both HOCS+ and HSCO+ could also be formed via the following ion-molecule reactions:
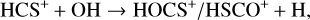 (1)
(1)
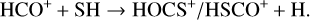 (2)
(2)
However, reactions (1) and (2) lack theoretical or experimental data on their rate coefficients, which prevents us from achieving a complete picture of the chemistry of their products and from determining whether a dominant formation of HOCS+ over HSCO+ does occur in the ISM. Therefore, the aim of this work is to study the thermodynamic and kinetic feasibility of these two reactions, using a combination of quantum chemistry and kinetic calculations.
The importance of studying these reactions is further supported by the widespread presence and astrochemical interest of the primary precursors, HCS+ and HCO+. Since its first detection toward the star-forming region Sgr B2 (Thaddeus et al. 1981), HCS+ has been detected in a wide variety of environments: translucent molecular clouds; cold molecular clouds; photo-dominated regions; the gas environment around protostellar objects; and the high-mass star-forming region in the Galactic disk, in the outer Galaxy, and also in external galaxies (Turner 1996; Leurini et al. 2006; Muller et al. 2013; Potapov et al. 2016; Fuente et al. 2016; Martín et al. 2021; Fontani et al. 2022, 2023). Moreover, in G+0.693 HCS+ is ∼5 times more abundant than HOCS+, further supporting its role as plausible precursor (Sanz-Novo et al. 2024a). In the case of HCO+, this O-bearing cation is even more ubiquitous (see e.g., Buhl & Snyder 1970; Klemperer 1970; Woods et al. 1975; Snyder et al. 1976; Hollis et al. 1976; Stark & Wolff 1979; Guelin et al. 1982; Ziurys et al. 2009; Najita et al. 2010; Pulliam et al. 2011; Hakobian & Crutcher 2012; Martín et al. 2021; Fontani et al. 2022).
In light of these observational results, it is also worth considering additional ion-molecule reactions involving chemically related species that could provide alternative formation pathways for HOCS+ and HSCO+. In particular, we also explore the following processes, which involve the high-energy isomer of protonated CO, HOC+, and the radical HCO:
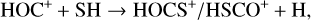 (3)
(3)
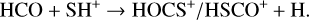 (4)
(4)
2 Computational methods
The formation processes analyzed in this study were characterized using ab initio and density functional theory (DFT) methodologies. At the DFT level, we selected the doublehybrid B2PLYPD3 functional (Grimme 2006), which includes the Hartree-Fock exchange and a perturbative second-order correlation part, as well as Grimme’s D3BJ empirical dispersion term (Grimme et al. 2011). For these calculations, we chose the Dunning’s correlation-consistent triple-zeta basis sets, aug-cc-pVTZ, (Dunning, Jr. 1989; Kendall et al. 1992; Woon & Dunning 1993), which includes both polarization and diffuse functions on all elements. For ab initio calculations, we selected the explicitly correlated coupled cluster theory with single and double excitations, including triplet excitations through a perturbative treatment, CCSD(T)-F12b (Knizia et al. 2009), in conjunction with the cc-pVTZ-F12 basis set (Peterson et al. 2008).
In addition, a composite approach was employed to achieve maximum accuracy in the relative energies of the stationary points located on the potential energy surface (PES) for the reactions analyzed kinetically. The calculated energy at the ab initio CCSD(T)/aug-cc-pV(T+d)Z level, a coupled cluster with single and double excitations, including triple excitations through a perturbative treatment (Raghavachari et al. 1989; Dunning et al. 2001), is taken as the reference for this approach. The aug-cc-pV(T+d)Z adds high-exponent functions for the sulfur atom to the standard correlation-consistent basis sets. Composite energies were calculated according to the expression:
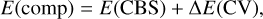
where E(CBS) corresponds to the complete basis set (CBS) limit extrapolated value, which considers basis set truncation errors, computed with an exponential extrapolation scheme (Halkier et al. 1999) using CCSD(T)/aug-cc-pV(X+d)Z (X = T, Q, and 5) data. The core-valence (CV) effects are considered through the term ∆E(CV). It is estimated as the difference between a calculation including all electrons (ae) and a frozen-core (fc) approach, computed using the Dunning’s weighted correlation-consistent polarized core-valence triple-zeta basis sets (aug-cc-pwCVTZ; Peterson & Dunning, Jr. 2002):
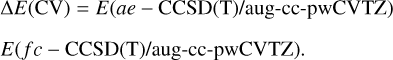
At the levels of calculation used in each optimized geometry, a calculation of harmonic vibrational frequencies was performed to characterize the stationary points and to determine the zeropoint vibrational energy (ZPVE). Critical points on the PES are classified as minima if they have all real vibrational frequencies, and transition states if they possess just one imaginary frequency. To confirm that transition states connect the desired minima, an intrinsic reaction coordinate (IRC) was calculated for each one (Fukui 1981).
For the most viable reaction from a thermodynamic point of view (see Section 3.1), we estimated rate constants and product distributions as a function of temperature and pressure by applying statistical rate theory to the stationary points using the Master Equation Solver for Multi-Energy well Reactions (MESMER) program (Glowacki et al. 2012). Theoretical determination of rate constants for multiwell reactions involves computing each elementary step individually. The transition state theory (TST) was employed to calculate the rate constants for the steps involving a transition state. Specifically, the microcanonical rate coefficient for these unimolecular elementary steps was determined using the Rice-Ramsperger-Kassel-Marcus (RRKM) theory (Holbrook et al. 1996; Baer & Hase 1996). The association rate coefficient corresponding to the entrance channels (barrierless steps) was calculated using Equation (5), proposed by Georgievskii and Klippenstein for ion-dipole interactions (Georgievskii & Klippenstein 2005):
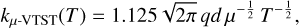 (5)
(5)
where q is the charge of the ion, d is the permanent dipole moment of the molecule, μ represents the reduced mass of the two reactants (in atomic mass unit), and T represents the temperature (in kelvin). This was then treated with the inverse Laplace transformation (ILT) methodology implemented in MESMER (Davies et al. 1986). This approximation resolves the challenge of explicitly defining a transition state for a barrierless process.
Electronic structure calculations were carried out using the GAUSSIAN 16 (Frisch et al. 2019) and MOLPRO (Werner et al. 2019) program packages, with methods, basis sets, and geometry optimization procedures implemented in them.
3 Results and discussion
In this section, we present and discuss the results of this work, beginning with the thermochemical analysis of the proposed gas-phase ion-molecule reactions that lead to the formation of protonated OCS isomers. For reactions identified as thermodynamically feasible under ISM conditions, the stationary points on their corresponding PESs are characterized. These data enable the determination of kinetic rate coefficients, providing insight into the relative formation ratios of the HOCS+ and HSCO+ isomers.
3.1 Formation of HOCS+ and HSCO+ isomers
In addition to studying reactions (1) and (2) proposed by Sanz-Novo et al. (2024a), we also considered processes involving HOC+, the radical HCO, and SH+ species (i.e., reactions (3) and (4)), all of which are known to be present in the ISM. Table 1 shows the reaction energies derived. In all reactions, we have considered both protonated isomers of OCS, HOCS+, and HSCO+ as possible products. As shown, for reaction (1) the formation of the sulfur-protonated isomer, HSCO+, is the most thermodynamically favorable process, while for reactions (2), (3), and (4), H2S+ is the predominant product, along with CO.
Reaction (1), HCS+ + OH, is exothermic for the formation of the two protonated isomers of OCS, while the competing proton transfer process, from HCS+ to OH, is clearly endothermic. In reaction (2), by contrast, the formation of either HOCS+ or HSCO+ follows endothermic pathways, while proton transfer, from HCO+ to SH, is clearly exothermic. Thus, proton transfer dominates, and reaction (2) does not contribute to the formation of the target species. On the other hand, for reactions (3) and (4), the formation of HOCS+ and HSCO+ is exothermic, but the process giving CO + H2S+ is energetically more favorable (see Table 1). A preliminary study shows that HOCS+ and HSCO+ are expected to be only minor products from the reactions HOC+ + SH, and HCO+ + SH, as can be seen in the PESs shown in Appendix A (Figs. A.1 and A.2). Furthermore, considering the relative abundances of the three precursors in G+0.693 (i.e., HCS+ is ∼2.8 and ∼22 times more abundant than HCO and HOC+, respectively; V.M. Rivilla, private communication), the only source where HOCS+ has been characterized so far, processes (3) and (4) are unlikely to play a significant role in the formation of HOCS+ and HSCO+.
Based on the aforementioned reactions and in view of current results, the formation of HOCS+ and HSCO+ is the only viable process from reaction (1). To see how both species are formed and determine their formation rate constants, it is necessary to characterize the critical points, intermediates, and transition states on the corresponding PES. We applied different calculation levels, described in Section 2, to study the PES of the reaction between OH and HCS+ (reaction (1)). Fig. 1 shows the reaction pathways that connect the localized stationary points. The relative energies for intermediates, products, and transition states computed using the reactants as a reference are listed in Table 2.
An analysis of the relative energies obtained at the different levels, summarized in Table 2, shows that, as expected, the results at the correlated CCSD(T)-F12b/cc-pVTZ-F12 level agree more closely with those from the composite calculations than those obtained at the CCSD(T)/aug-cc-pV(X+d)Z (X = T, Q, 5) levels. Except for intermediate I1, the discrepancies between the composite and the CCSD(T)-F12b/cc-pVTZ-F12 energy differences are below 0.4 kcal mol−1. At the B2PLYPD3/aug-cc-pVTZ level, the same qualitative trend is observed, but the energy differences relative to the composite values are larger (on the order of several kcal mol−1.
As shown in Table 2, all computational levels employed confirm that the sulfur-protonated OCS (HSCO+) is the most favorable product from a thermodynamic point of view. An analysis of the reaction pathways leading to both protonated isomers reveals that their formation takes place via the intermediate denoted as I1 (see Fig. 1). This intermediate can be reached through two different pathways. In the first path, I1a, the reaction begins with the interaction between HCS+ and OH, forming the ion-molecule complex C1. From this complex, C-O bond formation occurs through the transition state TS1, located 2.7 kcal mol−1 below the reactants at the composite level. The second path, I1b, starts with the formation of the ion-molecule complex C2, which evolves to intermediate I2 through the transition state TS2, where an S-O bond is formed. The TS2 transition state connects C2 with the I2 isomer in the “in” conformation, which subsequently evolves into the slightly more stable “out” conformer (by 0.2 kcal/mol). This interconversion process involves a barrier of 2.3 kcal/mol, which remains well below the energy of the reactants (−14.0 kcal/mol). Then, the migration of the OH group from sulfur to carbon in I2 leads to intermediate I1, via the transition state TS3. Both the TS2 and TS3 transition states are located below the reactants (0.9 and 8.0 kcal mol−1 at the composite level, respectively). These two paths (shown with dashed and dotted black lines), which give rise to intermediate I1, can be summarized as
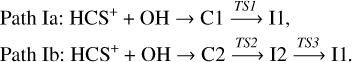
Once intermediate I1 is reached, the elimination of the hydrogen atom bonded to the carbon one leads to the formation of HOCS+. This process occurs through the transition state TS4, which is located at −7.6 kcal mol−1 at the composite level. This path, highlighted in green, can be summarized as follows:
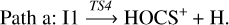
Alternatively, intermediate I1 can isomerize to intermediate I3 by migration of the hydrogen atom from carbon to sulfur, via transition state TS5, located 12.6 kcal mol−1 below the reactants. The two products, HOCS+ and HSCO+, can be obtained from isomer I3 by eliminating the hydrogen bonded to either the sulfur atom or the oxygen one, respectively. These processes involve passing through transition states TS6 and TS7, located at −11.7 and −3.5 kcal mol−1 from the reactants at the composite level. Both paths (shown in blue and purple) can be summarized as:
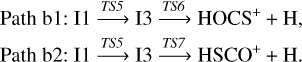
Based on the PES analysis, we can conclude that both isomers, HOCS+/HSCO+, can be formed from the reaction of the hydroxyl radical with HCS+. The barrier to the isomerisation of HSCO+ to HOCS+, calculated by Wheeler et al. (2006), lies ∼69 kcal/mol higher than the lower energy isomer. This high barrier hinders the isomerization process from taking place under ISM conditions; thus, if both isomers are indeed formed, they will coexist. To assess the efficiency of forming both products and to rationalize their observed abundances in the ISM, kinetic calculations are required.
Reaction energies including ZPVE calculated at different levels of theory (in kcal mol−1).
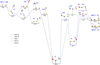 |
Fig. 1 Relative energies (in kcal mol−1) for the stationary points located along the gas phase reaction of HCS+ with hydroxyl radical computed at the composite approach. The ZPV energies are included. Different paths are highlighted with different line styles and colors, as shown in the legend. |
Relative energies including ZPVE for the stationary points located for the reaction between HCS+ and OH calculated at different levels of theory (in kcal mol−1).
Bimolecular rate coefficients, k(T), and branching ratios for the reaction of HCS+ with OH, computed at different temperatures with energies calculated at the composite approach and a density of 104 cm−3).
3.2 Rate coefficients for the HCS+ + OH reaction
The rate coefficients for the three pathways (a1, b1, and b2) of the HCS+ + OH reaction were calculated using the MESMER program. As input parameters, we used the composite relative energies (Table 2), along with the vibrational frequencies and rotational constants, of all species involved, computed at the CCSD(T)-F12b/cc-pVTZ-F12 level of theory (shown in Appendix B, Table B.1).
Initially, we performed simulations over a wide range of densities (104−1014 cm−3) and temperatures (75-300 K), to cover the hydrogen volume density and kinetic temperature of the Galactic center molecular cloud G+0.693 (i.e., H2 volume densities of a few times 104 cm−3 and Tkin = 70-140 K; Requena-Torres et al. 2006; Zeng et al. 2018; Colzi et al. 2024). We observed that the HCS+ + OH reaction exhibits no pressure dependence for the global rate constant within the investigated gas density range. Therefore, the gas density was set at 104 cm−3 , consistent with that of the gas in G+0.693. A minimum temperature of 75 K was selected since the MESMER program encounters difficulties in accurately predicting rate coefficients at low temperatures when the PES includes deep wells. This difficulty arises from the evaluation of both the forward and reverse reactions of these wells. At low temperatures, the rates out of the well become very low, often approaching the precision limits of the computation.
Table 3 presents the bimolecular global rate coefficients k1 and k2, for the formation of HOCS+ and HSCO+ respectively, from the reaction of HCS+ with OH. The rate coefficient k1 includes paths a and b1, whereas k2 corresponds to pathway b2. Table 3 also reports the branching ratios corresponding to the formation of HOCS+ and HSCO+.
As shown in Table 3, both k1 and k2 exhibit a negative temperature dependence over the temperature range considered in this study, consistent with the presence of negative-energy transition states on the PES for the reaction HCS+ with OH (Fig. 1). Furthermore, the global rate coefficient k1, associated with the formation of HOCS+, is significantly larger (i.e., by approximately two orders of magnitude) than k2, which corresponds to the formation of HSCO+. This behavior is in line with the lower activation barriers identified for pathways a and b1, in contrast to the higher barriers along pathway b2. The difference between the two rate coefficients is consequently reflected in the [HOCS+]/[HSCO+] ratios. Additionally, the branching ratios display a slight inverse linear dependence on temperature, increasing as the temperature decreases. As a result, the relative abundance of the less stable isomer, HOCS+, which is kinetically the most favorable, increases slightly at lower temperatures. Overall, these theoretical kinetic results indicate that the reaction preferentially proceeds via the mechanism leading to the less stable isomer, HOCS+, rather than to the more thermodynamically favorable product HSCO+.
4 Astrochemical implications and conclusions
The focus of recent astrochemical studies has recently shifted toward exploring the entire isomeric landscape of molecules, beyond merely the most stable isomer, as other high-energy isomers may also exist and be observable in space (see, e.g., recent interstellar detections of high-energy isomers within the CH2N, C3H2O, C2H4O2, C3H4O, and C2H5NO2 families; Loomis et al. 2015; Bermúdez et al. 2018; Mininni et al. 2020; Cabezas et al. 2021; Shingledecker et al. 2020; Rivilla et al. 2023; Sanz-Novo et al. 2025). The [H,C,S,O]+ isomeric family provides a further compelling case, in which HOCS+ has been detected despite being less stable (4.9 kcal mol−1 or 2456 K at the CCSD(T)/CBS level) than the global minimum, HSCO+, which is yet to be observed.
This finding, together with the lower limit isomeric ratio of HOCS+/HSCO+ ≥ 2.3 derived toward G+0.693 (Sanz-Novo et al. 2024a), provides strong observational evidence that thermodynamics alone cannot account for either the detectability or the relative abundance of these species. It adds to the growing list of exceptions to the minimum energy principle (MEP) outlined above, which proposed that the most thermodynamically stable isomer should be the most abundant in space (Lattelais et al. 2010). We must, therefore, invoke selective formation chemical routes, i.e., pathways that preferentially lead to the formation of the high-energy isomer (i.e., HOCS+ in this case), as proposed for other families, such as those yielding selectively trans methyl formate (CH3OCHO), cis formic acid (HCOOH), or glycolamide (HOCH2 C(O)NH2), a high-energy structural isomer of glycine, among others (Neill et al. 2012; García de la Concepción et al. 2022; Rivilla et al. 2023; Sanz-Novo et al. 2025; Perrero et al. 2025). In this context, the theoretical kinetic results presented here for reaction (1) suggest that this formation pathway is indeed favored for the production of HOCS+, which clearly predominates over HSCO+. While these results are in good agreement with the astronomical observations toward G+0.693, if the detection of HSCO+ is confirmed in the near future and the derived upper limit for HSCO+ is indeed close to its actual abundance, this will mean that the ratio of the rate constants (k1/k2) at a T ~ T kin (G+0.693) = 70-140 K is two orders of magnitude larger than the derived HOCS+/HSCO+ lower limit observational ratio (of ≥2.3) (Requena-Torres et al. 2006; Zeng et al. 2018; Colzi et al. 2024). Therefore, this scenario would suggest that additional formation pathways may also play a significant role in shaping the isomeric ratio.
Another key reaction is the direct protonation of OCS, previously suggested by Sanz-Novo et al. (2024a) as the main contributing pathway. While we cannot rule out a priori that this reaction may preferentially form the more stable isomer HSCO+, McCarthy & Thaddeus (2007) and Sanz-Novo et al. (2024a) proposed an alternative hypothesis based on chemical intuition. It suggests that the relative orientation of OCS and the protonating agent (e.g., H3+, HCO+, CH3+) might play a pivotal role in directing the direct protonation pathway. If the reaction depends on the spatial arrangement of the reactants, HOCS+ may dominate over HSCO+ in the ISM due to the higher electronegativity of O compared to S, making it the more favorable protonation site (McCarthy & Thaddeus 2007), again in agreement with observations toward G+0.693.
In addition to finding that reaction (1) is isomer-dependent, the relevance of this work to understanding the formation of HOCS+ lies in the fact that reaction (2) - proposed by Sanz-Novo et al. (2024a) - is endothermic for both the O- and S-protonated isomers and therefore inactive under interstellar conditions. Although reactions (3) and (4) proceed exothermically, their primary outcome is the formation of CO + H2S+, with HOCS+ and HSCO+ arising only as minor by-products. Thus, these routes can be disregarded as predominant formation channels of either HOCS+ or HSCO+.
In summary, we have found that the high-energy isomer HOCS+ is preferentially formed via the reaction of HCS+ and OH, which helps us rationalize its recent discovery in the ISM, along with the non-detection of HSCO+ toward G+0.693. We emphasize the importance of exploring the full isomeric panorama within a given family from both an experimental (i.e., spectroscopic and kinetic investigations) and theoretical point of view. However, quantum chemical computations on the reactivity and formation mechanisms of interstellar systems often overlook the full isomeric landscape. Incorporating all isomers, along with their specific formation pathways, into astrochemical models-many of which are still insensitive to isomerism-represents a critical step toward elucidating their formation routes and explaining and/or predicting their abundance ratios, which will be crucial to achieving a more complete picture of the chemistry of the targeted species in space.
Acknowledgements
The authors would like to thank Professor A. Fernández-Ramos for helpful discussions on kinetics calculations. V.M.R. acknowledges support from the grant RYC2020-029387-I funded by the Spanish Ministry of Science, Innovation and Universities/State Agency of ResearchMI-CIU/AEI/10.13039/501100011033 and by “ESF, Investing in your future”, and from the Consejo Superior de Investigaciones Científicas (CSIC) and the Centro de Astrobiología (CAB) through the project 20225AT015 (Proyectos intramurales especiales del CSIC); and from the grant CNS2023-144464 funded by MICIU/AEI/10.13039/501100011033 and by “European Union NextGenera-tionEU/PRTR”. M.S.-N. acknowledges a Juan de la Cierva Postdoctoral Fellow proyect JDC2022-048934-I, funded by MICIU/AEI/10.13039/501100011033 and by the European Union “NextGenerationEU”/PRTR”. M.S.-N. and V.M.R. acknowledge funding from the grant PID2022-136814NB-I00 by MICIU/AEI/10.13039/501100011033 and by ERDF, UE; and Consejo Superior de Investigaciones Científicas (CSIC) through project i-LINK23017 SENTINEL. C.B. and P.R. acknowledge support from the Spanish Ministerio de Ciencia e Innovación (PID2020-117742GBI00/AEI/10.13039/501100011033).
References
- Adams, N., Smith, D., & Grief, D. 1978, Int. J. Mass Spectrom. Ion Phys., 26, 405 [NASA ADS] [CrossRef] [Google Scholar]
- Baer, T., & Hase, W. 1996, Unimolecular Reaction Dynamics: Theory and Experiments (New York: Oxford University Press) [Google Scholar]
- Bermúdez, C., Tercero, B., Motiyenko, R. A., et al. 2018, A&A, 619, A92 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Boogert, A. C. A., Gerakines, P. A., & Whittet, D. C. B. 2015, ARA&A, 53, 541 [Google Scholar]
- Buhl, D., & Snyder, L. E. 1970, Nature, 228, 267 [NASA ADS] [CrossRef] [Google Scholar]
- Cabezas, C., Agúndez, M., Marcelino, N., et al. 2021, A&A, 654, A45 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Chang, Y., Fu, Y., Chen, Z., et al. 2023, Chem. Sci., 14, 8255 [Google Scholar]
- Colzi, L., Martin-Pintado, J., Zeng, S., et al. 2024, A&A, 690, A121 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Davies, J., Green, N., & Pilling, M. 1986, Chem. Phys. Lett., 126, 373 [Google Scholar]
- Dunning, Jr., T. H. 1989, J. Chem. Phys., 90, 1007 [Google Scholar]
- Dunning, T. H., Peterson, K. A., & Wilson, A. K. 2001, J. Chem. Phys., 114, 9244 [NASA ADS] [CrossRef] [Google Scholar]
- Fontani, F., Vagnoli, A., Padovani, M., et al. 2018, MNRAS, 481, L79 [NASA ADS] [CrossRef] [Google Scholar]
- Fontani, F., Colzi, L., Bizzocchi, L., et al. 2022, A&A, 660, A76 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Fontani, F., Roueff, E., Colzi, L., & Caselli, P. 2023, A&A, 680, A58 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., et al. 2019, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT [Google Scholar]
- Fuente, A., Cernicharo, J., Roueff, E., et al. 2016, A&A, 593, A94 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Fukui, K. 1981, Acc. Chem. Res., 14, 363 [CrossRef] [Google Scholar]
- García de la Concepción, J., Colzi, L., Jiménez-Serra, I., et al. 2022, A&A, 658, A150 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Georgievskii, Y., & Klippenstein, S. J. 2005, J. Chem. Phys., 122, 194103 [Google Scholar]
- Glowacki, D. R., Liang, C.-H., Morley, C., Pilling, M. J., & Robertson, S. H. 2012, J. Phys. Chem. A, 116, 9545 [NASA ADS] [CrossRef] [Google Scholar]
- Goldsmith, P. F., & Linke, R. A. 1981, ApJ, 245, 482 [NASA ADS] [CrossRef] [Google Scholar]
- Grimme, S. 2006, J. Chem. Phys., 124, 034108 [Google Scholar]
- Grimme, S., Ehrlich, S., & Goerigk, L. 2011, J. Computat. Chem., 32, 1456 [Google Scholar]
- Guelin, M., Cernicharo, J., & Linke, R. A. 1982, ApJ, 263, L89 [CrossRef] [Google Scholar]
- Gómez Martín, J. C., Brooke, J. S. A., Feng, W., et al. 2017, J. Geophys. Res.: Atmos., 122, 7678 [Google Scholar]
- Hakobian, N. S., & Crutcher, R. M. 2012, ApJ, 758, L18 [Google Scholar]
- Halkier, A., Helgaker, T., Jørgensen, P., Klopper, W., & Olsen, J. 1999, Chem. Phys. Lett., 302, 437 [Google Scholar]
- Harada, N., Martín, S., Mangum, J. G., et al. 2022, ApJ, 938, 80 [NASA ADS] [CrossRef] [Google Scholar]
- Holbrook, K., Pilling, M., & Robertson, S. 1996, Unimolecular Reactions, 2nd edn. (Chichester: Wiley) [Google Scholar]
- Hollis, J. M., Snyder, L. E., Lovas, F. J., & Buhl, D. 1976, ApJ, 209, L83 [NASA ADS] [CrossRef] [Google Scholar]
- Kendall, R. A., Jr., T. H. D., & Harrison, R. J. 1992, J. Chem. Phys., 96, 6796 [NASA ADS] [CrossRef] [Google Scholar]
- Klemperer, W. 1970, Nature, 227, 1230 [Google Scholar]
- Knizia, G., Adler, T. B., & Werner, H.-J. 2009, J. Chem. Phys., 130, 054104 [Google Scholar]
- Krasnopolsky, V. A. 2012, Icarus, 218, 230 [NASA ADS] [CrossRef] [Google Scholar]
- Lattanzi, V., Spezzano, S., Laas, J. C., et al. 2018, A&A, 620, A184 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Lattanzi, V., Sanz-Novo, M., Rivilla, V. M., et al. 2024, A&A, 689, A260 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Lattelais, M., Pauzat, F., Ellinger, Y., & Ceccarelli, C. 2010, A&A, 519, A30 [CrossRef] [EDP Sciences] [Google Scholar]
- Leurini, S., Rolffs, R., Thorwirth, S., et al. 2006, A&A, 454, L47 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Li, J., Wang, J., Zhu, Q., Zhang, J., & Li, D. 2015, ApJ, 802, 40 [Google Scholar]
- Liu, J., Van Devener, B., & Anderson, S. L. 2002, J. Chem. Phys., 117, 8292 [Google Scholar]
- Loomis, R. A., McGuire, B. A., Shingledecker, C., et al. 2015, ApJ, 799, 34 [Google Scholar]
- Majumdar, L., Gratier, P., Wakelam, V., et al. 2018, MNRAS, 477, 525 [NASA ADS] [CrossRef] [Google Scholar]
- Martín, S., Mangum, J. G., Harada, N., et al. 2021, A&A, 656, A46 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- McCarthy, M. C., & Thaddeus, P. 2007, J. Chem. Phys., 127, 221104 [Google Scholar]
- Mininni, C., Beltrán, M. T., Rivilla, V. M., et al. 2020, A&A, 644, A84 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Muller, S., Beelen, A., Black, J. H., et al. 2013, A&A, 551, A109 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Najita, J. R., Carr, J. S., Strom, S. E., et al. 2010, ApJ, 712, 274 [NASA ADS] [CrossRef] [Google Scholar]
- Neill, J. L., Muckle, M. T., Zaleski, D. P., et al. 2012, ApJ, 755, 153 [Google Scholar]
- Perdigon, J., de Laverny, P., Recio-Blanco, A., et al. 2021, A&A, 647, A162 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Perrero, J., Alessandrini, S., Ye, H., Puzzarini, C., & Rimola, A. 2025, A&A, 698, A51 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Peterson, K. A., & Dunning, Jr., T. H. 2002, J. Chem. Phys., 117, 10548 [Google Scholar]
- Peterson, K. A., Adler, T. B., & Werner, H.-J. 2008, J. Chem. Phys., 128, 084102 [Google Scholar]
- Potapov, A., Sánchez-Monge, Á., Schilke, P., et al. 2016, A&A, 594, A117 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Pulliam, R. L., Edwards, J. L., & Ziurys, L. M. 2011, ApJ, 743, 36 [NASA ADS] [CrossRef] [Google Scholar]
- Raghavachari, K., Trucks, G. W., Pople, J. A., & Head-Gordon, M. 1989, Chem. Phys. Lett., 157, 479 [Google Scholar]
- Rakshit, A. B. 1982, Int. J. Mass Spectrom. Ion Process., 41, 185 [Google Scholar]
- Requena-Torres, M. A., Martín-Pintado, J., Rodríguez-Franco, A., et al. 2006, A&A, 455, 971 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Rey-Montejo, M., Jiménez-Serra, I., Martín-Pintado, J., et al. 2024, ApJ, 975, 174 [NASA ADS] [CrossRef] [Google Scholar]
- Richardson, C. D., Hinman, N. W., & Scott, J. R. 2013, Int. J. Astrobiol., 12, 357 [Google Scholar]
- Rivilla, V. M., Jiménez-Serra, I., Martín-Pintado, J., et al. 2022, Front. Astron. Space Sci., 9 [Google Scholar]
- Rivilla, V. M., Sanz-Novo, M., Jiménez-Serra, I., et al. 2023, ApJ, 953, L20 [NASA ADS] [CrossRef] [Google Scholar]
- Rodríguez-Almeida, L. F., Jiménez-Serra, I., Rivilla, V. M., et al. 2021, ApJ, 912, L11 [Google Scholar]
- Sanz-Novo, M., Rivilla, V. M., Jiménez-Serra, I., et al. 2024a, ApJ, 965, 149 [Google Scholar]
- Sanz-Novo, M., Rivilla, V. M., Müller, H. S. P., et al. 2024b, ApJ, 965, L26 [Google Scholar]
- Sanz-Novo, M., Molpeceres, G., Rivilla, V. M., & Jimenez-Serra, I. 2025, A&A, 698, A36 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Sanz-Novo, M., Rivilla, V. M., Endres, C. P., et al. 2025, ApJ, 980, L37 [Google Scholar]
- Shingledecker, C. N., Molpeceres, G., Rivilla, V. M., Majumdar, L., & Kästner, J. 2020, ApJ, 897, 158 [NASA ADS] [CrossRef] [Google Scholar]
- Smith, D., & Adams, N. 1978, Chem. Phys. Lett., 54, 535 [Google Scholar]
- Snyder, L. E., Hollis, J. M., Lovas, F. J., & Ulich, B. L. 1976, ApJ, 209, 67 [Google Scholar]
- Stark, A. A., & Wolff, R. S. 1979, ApJ, 229, 118 [NASA ADS] [CrossRef] [Google Scholar]
- Thaddeus, P., Guelin, M., & Linke, R. A. 1981, ApJ, 246, L41 [NASA ADS] [CrossRef] [Google Scholar]
- Tinacci, L., Pantaleone, S., Maranzana, A., et al. 2021, ApJS, 256, 35 [Google Scholar]
- Todd, Z. R. 2022, Life, 12, 1268 [Google Scholar]
- Turner, B. E. 1996, ApJ, 461, 246 [Google Scholar]
- Turner, B. E., Amano, T., & Feldman, P. A. 1990, ApJ, 349, 376 [NASA ADS] [CrossRef] [Google Scholar]
- Vidal, T. H. G., Loison, J.-C., Jaziri, A. Y., et al. 2017, MNRAS, 469, 435 [Google Scholar]
- Werner, H., Knowles, P., Knizia, G., et al. 2019, Cardiff, UK [Google Scholar]
- Wheeler, S. E., Yamaguchi, Y., & Schaefer, Henry F. I. 2006, J. Chem. Phys., 124, 044322 [Google Scholar]
- Woods, R. C., Dixon, T. A., Saykally, R. J., & Szanto, P. G. 1975, Phys. Rev. Lett., 35, 1269 [Google Scholar]
- Woon, D. E., & Dunning, T. H. 1993, J. Chem. Phys., 98, 1358 [NASA ADS] [CrossRef] [Google Scholar]
- Zeng, S., Jiménez-Serra, I., Rivilla, V. M., et al. 2018, MNRAS, 478, 2962 [Google Scholar]
- Ziurys, L. M., Tenenbaum, E. D., Pulliam, R. L., Woolf, N. J., & Milam, S. N. 2009, ApJ, 695, 1604 [Google Scholar]
The structural isomers HOCS+, HSCO+, and c-OC(H)S+ contain the same number of atoms of each element but differ in the arrangement of their atomic bonds. The latter cyclic species lies at 56.05 kcal mol−1 (30 219 K) above HSCO+ (Wheeler et al. 2006), yet it remains undetected in the laboratory.
Appendix A Complementary figures
Schematic representation of the stationary points characterized on the PESs for the HOC+ + SH (3) and HCO + SH+ (4) reactions, computed at the B2PLYPD3/aug-cc-pVTZ level.
 |
Fig. A.1 Relative energies (in kcal mol−1) for the stationary points located along the gas phase reaction of HOC+ and SH computed at the B2PLYPD3 level. The ZPV energies are included. From intermediate I3, HCOS+ and HSCO+ are obtained following the steps shown in Fig. 1. |
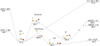 |
Fig. A.2 Relative energies (in kcal mol−1) for the stationary points located along the gas phase reaction of HCO + SH+ computed at the B2PLYPD3 level. The ZPV energies are included. From intermediate I3, HCOS+ and HSCO+ are obtained following the steps shown in Fig. 1. |
Appendix B Complementary table
We provide a list with the theoretical rotational constants and harmonic vibrational frequencies for the stationary points located on the doublet PES of the reaction between HCS+ and OH.
Theoretically predicted rotational constants, Ae, Be, and Ce, (in GHz) and harmonic vibrational frequencies, vi (in cm−1) for the stationary points located on the PES of the reaction between HCS+ and OH computed at the CCSD(T)-F12b/cc-pVTZ-F12 level.
All Tables
Reaction energies including ZPVE calculated at different levels of theory (in kcal mol−1).
Relative energies including ZPVE for the stationary points located for the reaction between HCS+ and OH calculated at different levels of theory (in kcal mol−1).
Bimolecular rate coefficients, k(T), and branching ratios for the reaction of HCS+ with OH, computed at different temperatures with energies calculated at the composite approach and a density of 104 cm−3).
Theoretically predicted rotational constants, Ae, Be, and Ce, (in GHz) and harmonic vibrational frequencies, vi (in cm−1) for the stationary points located on the PES of the reaction between HCS+ and OH computed at the CCSD(T)-F12b/cc-pVTZ-F12 level.
All Figures
|
Fig. 1 Relative energies (in kcal mol−1) for the stationary points located along the gas phase reaction of HCS+ with hydroxyl radical computed at the composite approach. The ZPV energies are included. Different paths are highlighted with different line styles and colors, as shown in the legend. |
| In the text | |
|
Fig. A.1 Relative energies (in kcal mol−1) for the stationary points located along the gas phase reaction of HOC+ and SH computed at the B2PLYPD3 level. The ZPV energies are included. From intermediate I3, HCOS+ and HSCO+ are obtained following the steps shown in Fig. 1. |
| In the text | |
|
Fig. A.2 Relative energies (in kcal mol−1) for the stationary points located along the gas phase reaction of HCO + SH+ computed at the B2PLYPD3 level. The ZPV energies are included. From intermediate I3, HCOS+ and HSCO+ are obtained following the steps shown in Fig. 1. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.