| Issue |
A&A
Volume 708, April 2026
|
|
|---|---|---|
| Article Number | A19 | |
| Number of page(s) | 6 | |
| Section | Atomic, molecular, and nuclear data | |
| DOI | https://doi.org/10.1051/0004-6361/202659186 | |
| Published online | 26 March 2026 | |
Isotopic effects in rotational de-excitation of H2CO isotopologues (HDCO and D2CO) by He at low temperatures
1
Departamento de Física, Facultad de Ciencias, Universidad de Chile,
Av. Las Palmeras 3425,
7800003
Ñuñoa,
Santiago,
Chile
2
Departamento de Física, Facultad de Ciencias, Universidad de Chile,
Cedenna, Av. Las Palmeras 3425,
7800003
Ñuñoa,
Santiago,
Chile
★ Corresponding authors: This email address is being protected from spambots. You need JavaScript enabled to view it.
; This email address is being protected from spambots. You need JavaScript enabled to view it.
; This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
28
January
2026
Accepted:
24
February
2026
Abstract
Context. In typical interstellar molecular clouds, densities are low; therefore, the analysis of observed molecular spectra requires using non-LTE models. Critical inputs for these models are collisional rate coefficients of the detected molecule with He and H2. For formaldehyde (H2CO), such data are available; however, no collisional data have been reported for its deuterated isotopologues, HDCO and D2CO.
Aims. The main goal of this work is to study isotopic effects in the collision of formaldehyde with He, particularly on HDCO and D2CO, and to report new sets of collisional rate coefficients for these systems at low temperatures.
Methods. New potential energy surfaces were developed for the H2CO+He and HDCO+He systems using ab initio calculations at the CCSD(T)-F12a/aug-cc-pVTZ level of theory. The H2CO+He potential energy surface was also shifted to the centre of mass of D2CO to study its collision with He. These potential energy surfaces were employed in close-coupling scattering calculations.
Results. The potential energy surfaces for H2CO+He and HDCO+He show differences that arise mainly from symmetry effects, leading to additional non-zero expansion coefficients in the HDCO+He case. The cross sections and rate coefficient are computed for para-H2CO, ortho-H2CO, para-D2CO, ortho-D2CO, and HDCO by collision with He. A comparison with available collisional data for H2CO+He is presented. Similarities in the collisional rate coefficients are found for the same transition for the three isotopologues. However, rotational de-excitation rate coefficients for transitions forbidden in H2CO+He but allowed in HDCO+He are reported for the first time.
Key words: astrochemistry / molecular data / molecular processes / scattering / ISM: molecules
© The Authors 2026
 Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Open Access article, published by EDP Sciences, under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article is published in open access under the Subscribe to Open model. This email address is being protected from spambots. You need JavaScript enabled to view it. to support open access publication.
1 Introduction
Typical conditions of interstellar molecular clouds are far from equilibrium due to their low densities (ranging from 102 to 108 particles per cubic centimetre; (Flower 2007)). Therefore, the analysis of observed spectra and, consequently, the accurate estimation of column densities, kinetic temperature, and gas volume density in molecular clouds must be performed using non-local thermodynamic equilibrium (non-LTE) models (van der Tak et al. 2020). Such models require knowledge of the Einstein coefficients and accurate state-to-state inelastic collisional rate coefficients of the observed molecules with the most common colliders in the interstellar medium (e.g. He, H, H2, and e−). The Einstein coefficients are usually known, but the collisional rate coefficients are not.
In cases where collisional data with H2 are not available for a given system, it is common to estimate the rates for collisions with H2 from those with He using a reduced-mass scaling factor (Schöier et al. 2005). If rates with He are also unavailable, scaling factors based on similar systems (e.g. isoelectronic systems) are used. For isotopologues, the same collisional data as for the parent species are usually used, except in the case of H/D substitution, for which the mass correction is larger than about 30% (van der Tak et al. 2020), and therefore cannot always be treated reliably by simple scaling.
Furthermore, astronomers commonly use deuterium fractionation (D/H ratio) derived from molecular abundance ratios to infer formation conditions. A high D/H ratio generally indicates a low formation temperature, and vice versa (Persson et al. 2018). Therefore, collisional data are required to estimate the abundances of hydrogenated and deuterated molecules.
When dealing with a deuterated isotopologue of a molecule, the centre of mass shifts, the principal inertia axes may rotate, the reduced mass of the complex changes, and molecular symmetries can be broken. As a result, the effect of H/D substitution on collisional rate coefficients is not straightforward.
In some cases, the H/D substitution has a limited impact on the rate coefficients, such as for HCO+/DCO+ in collisions with He (Buffa 2012; Buffa et al. 2009) or with H2 (Denis-Alpizar et al. 2020), and for N2H+/N2D+ + He (Daniel et al. 2007). However, in other systems, the influence of isotopic substitution is more pronounced. For example, in a recent study of ArH+/ArD+ in collisions with He (García-Vázquez et al. 2019), the rate coefficients show a different temperature dependence, indicating that a simple mass scaling does not allow one to estimate one set of rates from the other. Similar behaviour was observed for collisions of CH+/CD+ with He (Werfelli et al. 2017), C2H/C2D with para-H2 (Dumouchel et al. 2017), and for D2S–H2, whose cross sections were found to be larger than those of H2S–H2 (Dagdigian 2022).
Even larger discrepancies between the rate coefficients of isotopologues have been reported in some systems. This is the case for NH/ND in collisions with He (Dumouchel et al. 2012), as well as H2O/D2O (Scribano et al. 2010), with differences of up to a factor of 8, and HCN/DCN in collisions with He (Denis-Alpizar et al. 2015), where differences of up to a factor of 4 were found.
Additionally, there are H/D substitutions, for which symmetry breaking appears, as in the case of H2/HD (Roueff & Flower 1999; Flower 1999). Since the homonuclear symmetry breaking in HD allows transitions with odd ∆ j that are not permitted for H2, the comparison is not straightforward. Another example is H2O/HDO in collisions with He (Green 1989), where differences of about one order of magnitude were reported. For the same substitution in collisions with H2, Wiesenfeld et al. (2011) also found significant discrepancies. These works clearly show that specific calculations of collisional rate coefficients for deuterated isotopologues are necessary, as it is generally not possible to predict a priori the effect of H/D substitution on the collisional data.
Formaldehyde (H2CO) is one of the most abundant organic molecules in the interstellar medium and is widely used to probe the physical conditions of molecular gas (Maret et al. 2004). Its deuterated isotopologues, HDCO and D2CO, have also been detected (Persson et al. 2018). While collisional rate coefficients for H2CO with H2 (Wiesenfeld & Faure 2013) and with He (Green 1991) are available, no such data exist for its deuterated isotopologues. Significant differences are nevertheless expected, particularly for HDCO, for which symmetry is broken.
The main goals of this work are to study isotopic effects in the collision of formaldehyde with He, with a focus on HDCO and D2CO, and to report new sets of collisional rate coefficients for these systems at low temperatures. The paper is organized as follows. Section 2 presents the methods used to develop the potential energy surfaces (PES) and to perform the scattering calculations. Section 3 discusses the results, and Section 4 summarizes the main conclusions.
2 Methodology
2.1 Ab initio calculations
In this work, formaldehyde and its deuterated isotopologues are treated as rigid rotors. This approximation is excellent for collisional energies lower than the energy of the lowest vibrational excitation in triatomic systems colliding with atoms (Stoecklin et al. 2013, 2015; Cabrera-González et al. 2022). As we are interested in low temperatures and, therefore, low collision energies, this approximation is well justified for the present study (e.g. the lower frequency is ***(eq1)***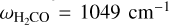 ; (Linstrom & Mallard 2025)).
; (Linstrom & Mallard 2025)).
Figure 1 shows the coordinate system used in this work. R is the distance between the centre of mass of the molecule and the He atom, θ is the angle between the z axis and the vector R, and φ is the corresponding azimuthal angle. In the case of HDCO, the molecule was rotated with respect to H2CO to ensure that the principal axes of inertia coincide with the Cartesian axes. In this deuterated case, the O–C bond forms an angle of 5.27◦ with respect to the z axis. The internal geometry of formaldehyde was fixed using the following values: the C–O bond length was set to 1.205 Å, the C–H bond length to 1.111 Å, the H–C–H bond angle to 116.1◦, and the H–C–O angle to 121.9◦ (Linstrom & Mallard 2025).
An analysis using different ab initio methods and basis sets was performed for the HDCO+He system. Figure 2 shows the interaction energies as a function of R computed at θ = 80◦ and φ = 0◦. In addition, interaction energies at the complete basis set limit (CBSL) were estimated from CCSD(T) calculations with the aug-cc-pVQZ and aug-cc-pV5Z basis sets using the two-point extrapolation formula (Helgaker et al. 1997)
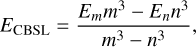
where Em and En are the interaction energies computed with basis sets of cardinal numbers m and n (with m = 5 and n = 4). Considering the computational cost and proximity to the CBSL energies, the CCSD(T)-F12a method with the aug-cc-pVTZ basis set was selected. The basis set superposition error was corrected using the counterpoise method (Boys & Bernardi 1970).
Two grids of interaction energies were computed, one for the H2CO+He complex and the other for the HDCO+He complex. Each grid includes 24 values of R ranging from 2.2 to 12 Å. The angle θ varies from 0◦ to 180◦ in steps of 10◦. The azimuthal angle φ was sampled from 0◦ to 180◦ in steps of 15◦ for HDCO, and from 0◦ to 90◦ for H2CO.
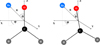 |
Fig. 1 Coordinate system used for the H2CO+He and HDCO+He complexes. In each case, the molecule lies in the x–z plane, with its centre of mass fixed at the origin. |
 |
Fig. 2 Interaction energies as a function of R , at θ = 80◦ and φ = 0◦, for the HDCO+He complex computed using CCSD(T) and CCSD(T)-F12 methods with different basis sets. The estimated CBSL energies are also shown for comparison. |
2.2 Analytical PES
The two grids of interaction energies were fitted to an analytical function of the form
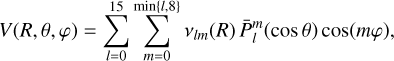 (1)
(1)
where ***(eq4)***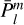 are the normalized associated Legendre polynomials. For the H2CO+He system, only even values of m were included owing to the C2v symmetry of the molecule.
are the normalized associated Legendre polynomials. For the H2CO+He system, only even values of m were included owing to the C2v symmetry of the molecule.
For a given value of R, the expansion coefficients νlm(R) were determined using singular value decomposition (SVD) (Press et al. 1992). For each pair (l, m), the corresponding radial coefficients were then fitted using a kernel-based method (Ho & Rabitz 1996), such that the radial dependence is written as
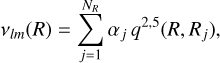 (2)
(2)
where NR is the number of radial grid points. The radial kernel is given by
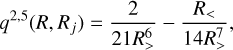 (3)
(3)
where R> (R<) denotes the larger (smaller) of R and Rj. The coefficients αj were obtained by solving the set of linear equations for each pair (l, m),
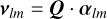
where Qij = q2,5(Ri, Rj) and Ri and Rj are the i and j radial points of the grid.
The potential energy surface for the D2CO+He complex was obtained by shifting the centre of mass of the H2CO+He surface, following the approach previously adopted for the D2O+Ar system based on the H2O+Ar surface (García-Vázquez et al. 2024).
MOLSCAT parameters used in the scattering calculations.
2.3 Scattering
The computed PESs were incorporated into the MOLSCAT code (Hutson & Le Sueur 2019). As the two hydrogen nuclei in the H2CO molecule are symmetrically equivalent fermions, the H2CO rotational energy levels are split into para (even Ka) and ortho (odd Ka) species (Green et al. 1978). This symmetry allows only para–para and ortho–ortho transitions. In the case of D2CO, the rotational levels are also split into para- and ortho-species. However, since deuterium is a boson with nuclear spin 1, the spin statistics assign para levels to odd Ka values and ortho levels to even Ka values, in contrast to the H2CO case (Turner 1990).
The inelastic cross sections were computed using the close-coupling method and the log-derivative propagator. The molecular rotational constants (A, B, C), centrifugal distortion constants (DJ, DJK, DK), reduced mass (URED), and other MOLSCAT parameters used in the calculations are summarized in Table 1. For all complexes, the scattering calculations employed RMIN = 2 Å, RMAX = 20 Å, and OTOL = 0.001 Å2.
The rotational basis sets were selected based on convergence tests. Convergence of the inelastic cross sections within a few percent was achieved using an energy cutoff of Emax = 400 cm−1 for the para-H2CO+He, para-D2CO+He, and ortho-D2CO+He complexes. For ortho-H2CO+He, convergence for the transitions of interest was already reached at Emax = 200 cm−1. In the case of HDCO+He, where symmetry breaking leads to a higher density of coupled rotational states, an intermediate value of Emax = 300 cm−1 was adopted.
The cross sections for D2CO and HDCO in collision with He were computed for initial rotational levels with energies up to approximately 53 cm−1. For the H2CO+He system, cross sections were computed only up to the first six rotational levels to compare with available data, since collisional rate coefficients for this system are already available in the literature. Calculations for the para and ortho species of H2CO and D2CO were performed independently. The number of initial rotational levels considered in each case is smaller than for HDCO+He. The total energy grids were chosen to ensure a denser sampling in the resonance region. Finally, the state-to-state rate coefficients were obtained by thermal averaging of the cross sections over a Maxwell–Boltzmann distribution of collision energies.
Global minimum of the PES for the H2CO+He complex (in cm−1).
 |
Fig. 3 First radial coefficients νlm(R) of the expansion PES for H2CO+He (solid line), HDCO+He (dashed lines), and D2CO+He (dotted line). |
 |
Fig. 4 Contour plots for the H2CO + He (top panels) and HDCO + He (bottom panels) complexes at selected configurations. Positive energies are shown in red contour lines, while negative energies are shown in blue lines in steps of 3 cm−1. |
3 Results
3.1 PES
The quality of the fitting of the PES was assessed through the root-mean-square deviation (RMSD) between the ab initio interaction energies and those obtained from the analytical representation given in Eq. (1). For the energy grid employed in the construction of the HDCO+He PES, the RMSD was 2.9 × 10−2 cm−1 for negative energies and 7.5 cm−1 for energies from 0 to 5000 cm−1. In addition, 432 off-grid ab initio points were selected to validate the analytical surface. For these points, the RMSD was 3.3 × 10−2 cm−1 in the attractive region (219 points) and 36.4 cm−1 in the repulsive region, where the energies extend up to 104 cm−1.
For the H2CO+He PES, the RMSD was 1.8 × 10−2 cm−1 for E < 0, while for energies between 0 and 5000 cm−1 it increases to 25.6 cm−1. For this system, several PESs are available in the literature. The global minimum obtained in the present work (−53.0 cm−1) is compared with previous results in Table 2. The agreement in the geometry of the global minimum is good. However, noticeable discrepancies are observed among the reported well depths.
The difference with the early work of Garrison & Lester Jr (1975) is not unexpected, given the substantially lower level of electronic structure theory employed in that study. However, the discrepancy with more recent PESs requires closer analysis. Wheeler & Ellis (2003) reported a global minimum of −59.5 cm−1 for the H2CO+He interaction, based on a PES fitted to CCSD(T) interaction energies extrapolated to the complete basis set limit (CBS) using aug-cc-pVDZ and aug-cc-pVTZ basis sets. In the same work, one-dimensional cuts computed at the CCSD(T)/aug-cc-pVQZ level show a noticeably shallower well. To understand such a difference, we computed the ab initio interaction energy at the geometry of the minimum reported by Wheeler & Ellis (2003). Using the standard two-point extrapolation formula, we obtain a value of −50.4 cm−1, in close agreement with our PES. However, applying the extrapolation expression (Eq. (1)) of Wheeler & Ellis (2003) yields a significantly deeper value of −57.7 cm−1. It is worth noting that this extrapolated value is considerably deeper than the interaction energy obtained directly with the aug-cc-pV5Z basis set (−49.22 cm−1). This behaviour suggests that the CBS extrapolation scheme employed, which includes two empirical parameters, may overestimate the interaction energy in the well region.
In the case of the PES reported by Naumkin et al. (2019), the difference with our results can be attributed to a combination of two factors. First, as shown in Fig. 2, the CCSD(T)-F12a/cc-pVQZ-F12 level underestimates the magnitude of the interaction energy for this system. Second, the fitting strategy adopted in that work relies on a relatively limited number of ab initio energy configurations, which may introduce additional smoothing of the potential near the global minimum.
The first radial coefficients of the expansion of the PESs are shown in Figure 3. There is an agreement between the coefficients of H2CO+He and D2CO+He, as expected, because the second PES is obtained from the first one simply by shifting the centre of mass by 0.07 Å. Furthermore, similarities are found between the νl,m terms for HDCO+He and H2CO+He for the even values of m. However, additional νl,m terms appear for HDCO+He (odd values of m), which are zero for H2CO+He and D2CO+He.
Figure 4 shows the contour plot for the HDCO+He and H2CO+He complexes. The D2CO+He plots are not shown because the difference with those of H2CO+He is so slight that it cannot be identified from a figure. The global minimum of both complexes is in the coplanar configuration, φ = 0◦, where for HDCO+He it is −53.0 at R = 3.18 Å and θ = 77.3◦, very close to the values of H2CO+He. The other minima also exhibit a slight displacement between the two systems.
The differences in the PESs of HDCO+He and H2CO+He are shown in more detail in Figure 5. In this figure, R was relaxed in the [2.5, 5.0] Å interval, and highlights the angular anisotropy of the PESs. The global minimum in both complexes is centred around θ ≈ 90◦ and ϕ ≈ 0◦, corresponding to a near T-shaped, coplanar configuration with He close to the O atom. Shallower minima are also observed for axial approaches towards the H–H side (θ ≳ 150◦) and the O end (θ ≲ 30◦). The interaction becomes less attractive as ϕ leaves the coplanar geometry. Furthermore, the dashed red line at φ = 90◦ highlights the breaking of the angular symmetry of the mass distribution in the HDCO+He complex.
 |
Fig. 5 Contour plots of the potential energy surface for the H2CO+He (top panel) and HDCO+He (bottom panel) complexes with R relaxed in the [2.5, 5.0] Å interval. The contour lines are in steps of 2 cm−1. |
 |
Fig. 6 Rotational de-excitation cross sections as a function of the collision energy for transitions from the 30,3 state. Top panel: para-H2CO+He (solid lines) and ortho-D2CO+He (dotted lines). Bottom panel: HDCO+He. |
 |
Fig. 7 Rotational de-excitation rate coefficients (solid lines) as a function of temperature for rotational transitions of para-H2CO (top panel) and ortho-H2CO (bottom panel). Data from Green (1991) (dash–dotted lines), and the mass-scaled data of Wiesenfeld & Faure (2013) (dashed lines) are also included. |
3.2 Scattering
The developed surfaces were incorporated into the MOLSCAT code to study the scattering of these systems. Figure 6 shows the rotational de-excitation cross sections as a function of the collision energy (Ecol) for H2CO, D2CO, and HDCO colliding with He. The typical shape and Feshbach resonances of this type of system are shown in this figure. These resonances are due to the temporary trapping of the collision complex behind centrifugal barriers associated with the van der Waals interaction, and to the coupling between open scattering channels and closed channels associated with rotationally excited levels of the formaldehyde isotopologues.
Furthermore, from the top panel of Figure 6, and as expected, it can be seen that the differences between transitions for H2CO and D2CO by collision with He are small. However, and as shown in the bottom panel, the 30,3 → 11,1, 30,3 → 11,0, 30,3 → 21,2, and 30,3 → 21,1 transitions do not appear for the H2CO–He and D2CO–He complexes but, are possible for the HDCO. This is because, in these formaldehyde molecules, transitions between para and ortho (and vice versa) are forbidden. In contrast, this symmetry restriction is lifted in the HDCO case, allowing these transitions.
Figure 7 shows selected de-excitation rate coefficients for the collision of H2CO with He. This figure includes the rate coefficients reported by Green (1991) (from the BASECOL database). For some transitions (e.g. 30,3 → 20,2, and 21,1 → 11,1 the agreement between (Green 1991) and the present work is excellent. However, in general, the differences between the rates are larger at low temperatures and decrease as T increases. This behaviour is not surprising, given the discrepancies in the PES employed by Green (1991), which was based on the surface of Garrison & Lester Jr (1975). Furthermore, the mass-scaled (Schöier et al. 2005) rate coefficients from the H2CO–H2 collisions of Wiesenfeld & Faure (2013), as available in the EMAA database, are also included in Figure 7. In this case, the rate coefficients are generally higher than our values, and a simple mass-scaling law appears inadequate for this system.
Rotational de-excitation rate coefficients for the collision of HDCO and D2CO with He are shown in Figure 8. For transitions allowed in D2CO+He, the rate coefficients for HDCO+He show a similar behaviour and magnitude. However, some transitions not allowed for H2CO+He and D2CO+He are permitted for HDCO+He; therefore, these transitions cannot be predicted simply from the corresponding data for H2CO+He and D2CO+He, making quantum calculations for this system necessary. The data computed for the first 25 rotational levels of HDCO and the first 16 rotational levels of para- and ortho-D2CO in collisions with He, up to 40 K, will be made available through the BASECOL database(Dubernet et al. 2024).
 |
Fig. 8 Rotational de-excitation rate coefficients from the initial state 30,3, for all the allowed rotational transitions of D2CO (dashed line) and HDCO (solid line) induced by collisions with He, plotted as a function of the temperature. |
4 Summary
The collisions of H2CO and its isotopologues, D2CO and HDCO, with He were studied in this work. New potential energy surfaces were developed from high-level ab initio calculations. For the H2CO+He system, for which previous studies are available, differences with earlier surfaces were found, mainly due to the level of theory employed in the electronic structure calculations. The PES for D2CO+He was obtained from that of H2CO+He by applying a correction to the centre of mass; therefore, the differences between these two surfaces are minor. In contrast, for the HDCO+He system, radial coefficients of the PES expansion that vanish for the symmetric isotopologues become significant.
The potential energy surfaces were employed in close-coupling scattering calculations, and five new sets of rotational de-excitation rate coefficients were computed, separating para and ortho species for H2CO and D2CO. The rate coefficients for transitions allowed in H2CO+He can be safely used in non-LTE calculations for D2CO+He collisions. However, for HDCO+He, additional rotational transitions are allowed that cannot be estimated from the symmetric isotopologues, confirming that rate coefficients must be computed independently for isotopologues lacking symmetry.
Data availability
The collisional rate coefficients for D2CO+He and HDCO+He will be available in the BASECOL database (Dubernet et al. 2024).
Acknowledgements
Support of projects Anid/Fondecyt Regular/ No1240102 and 1220366 is gratefully acknowledged. C.C. acknowledges support by Center for Nanoscience and Nanotechnology CEDENNA CIA250002. This research was partially supported by the supercomputing infrastructure of the NLHPC (CCSS210001).
References
- Boys, S. F., & Bernardi, F. 1970, Mol. Phys., 19, 553 [NASA ADS] [CrossRef] [Google Scholar]
- Buffa, G. 2012, MNRAS, 421, 719 [NASA ADS] [Google Scholar]
- Buffa, G., Dore, L., & Meuwly, M. 2009, MNRAS, 397, 1909 [Google Scholar]
- Cabrera-González, L. D., Denis-Alpizar, O., Páez-Hernández, D., & Stoecklin, T. 2022, MNRAS, 514, 4426 [Google Scholar]
- Dagdigian, P. J. 2022, MNRAS, 511, 3440 [NASA ADS] [CrossRef] [Google Scholar]
- Daniel, F., Cernicharo, J., Roueff, E., Gerin, M., & Dubernet, M. 2007, ApJ, 667, 980 [NASA ADS] [CrossRef] [Google Scholar]
- Denis-Alpizar, O., Stoecklin, T., & Halvick, P. 2015, MNRAS, 453, 1317 [CrossRef] [Google Scholar]
- Denis-Alpizar, O., Stoecklin, T., Dutrey, A., & Guilloteau, S. 2020, MNRAS, 497, 4276 [Google Scholar]
- Dubernet, M., Boursier, C., Denis-Alpizar, O., et al. 2024, A&A, 683, A40 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Dumouchel, F., Kłos, J., Toboła, R., et al. 2012, J. Chem. Phys., 137, 114306 [NASA ADS] [CrossRef] [Google Scholar]
- Dumouchel, F., Lique, F., Spielfiedel, A., & Feautrier, N. 2017, MNRAS, 471, 1849 [NASA ADS] [CrossRef] [Google Scholar]
- Flower, D. 1999, J. Phys. B: At. Mol. Opt. Phys., 32, 1755 [NASA ADS] [CrossRef] [Google Scholar]
- Flower, D. 2007, Molecular Ccollisions in the Interstellar Medium (Cambridge University Press), 42 [Google Scholar]
- García-Vázquez, R. M., Márquez-Mijares, M., Rubayo-Soneira, J., & Denis-Alpizar, O. 2019, A&A, 631, A86 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- García-Vázquez, R. M., Cabrera-González, L. D., Denis-Alpizar, O., & Stoecklin, T. 2024, Chem. Phys. Chem., 25, e202300752 [CrossRef] [Google Scholar]
- Garrison, B. J., & Lester Jr, W. A. 1975, J. Chem. Phys., 63, 4167 [Google Scholar]
- Green, S. 1989, ApJS, 70, 813 [NASA ADS] [CrossRef] [Google Scholar]
- Green, S. 1991, ApJS, 76, 979 [NASA ADS] [CrossRef] [Google Scholar]
- Green, S., Garrison, B. J., Lester, W. A., Jr., & Miller, W. H. 1978, ApJS, 37, 321 [Google Scholar]
- Helgaker, T., Klopper, W., Koch, H., & Noga, J. 1997, J. Chem. Phys., 106, 9639 [NASA ADS] [CrossRef] [Google Scholar]
- Ho, T. S., & Rabitz, H. 1996, J. Phys. Chem., 104, 2584 [CrossRef] [Google Scholar]
- Hutson, J. M., & Le Sueur, C. R. 2019, Comput. Phys. Commun., 241, 9 [NASA ADS] [CrossRef] [Google Scholar]
- Linstrom, P. J., & Mallard, W. G. 2025, NIST Chemistry WebBook, NIST Standard Reference Database Number 69, accessed on December, 2025 [Google Scholar]
- Maret, S., Ceccarelli, C., Caux, E., et al. 2004, A&A, 416, 577 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Müller, H. S., Schlöder, F., Stutzki, J., & Winnewisser, G. 2005, J. Mol. Struct., 742, 215 [Google Scholar]
- Naumkin, F., del Mazo-Sevillano, P., Aguado, A., Suleimanov, Y. V., & Roncero, O. 2019, ACS Earth Space Chem., 3, 1158 [Google Scholar]
- Persson, M. V., Jørgensen, J., Müller, H., et al. 2018, A&A, 610, A54 [NASA ADS] [CrossRef] [EDP Sciences] [Google Scholar]
- Press, W. H., Flannery, B. P., Teukolsky, S. A., & Vetterling, W. T. 1992, Numerical Recipes in FORTRAN 77: The Art of Scientific Computing, 2nd edn. (Cambridge: Cambridge University Press) [Google Scholar]
- Roueff, E., & Flower, D. 1999, MNRAS, 305, 353 [CrossRef] [Google Scholar]
- Schöier, F. L., van der Tak, F. F. S., van Dishoeck, E. F., & Black, J. H. 2005, A&A, 432, 369 [Google Scholar]
- Scribano, Y., Faure, A., & Wiesenfeld, L. 2010, J. Chem. Phys., 133, 231105 [Google Scholar]
- Stoecklin, T., Denis-Alpizar, O., & Halvick, P. 2015, MNRAS, 449, 3420 [NASA ADS] [CrossRef] [Google Scholar]
- Stoecklin, T., Denis-Alpizar, O., Halvick, P., & Dubernet, M.-L. 2013, J. Chem. Phys., 139, 034304 [NASA ADS] [CrossRef] [Google Scholar]
- Turner, B. E. 1990, ApJ, 362, L29 [Google Scholar]
- van der Tak, F. F., Lique, F., Faure, A., Black, J. H., & van Dishoeck, E. F. 2020, Atoms, 8, 15 [NASA ADS] [CrossRef] [Google Scholar]
- Werfelli, G., Balança, C., Stoecklin, T., Kerkeni, B., & Feautrier, N. 2017, MNRAS, 468, 2582 [Google Scholar]
- Wheeler, M. D., & Ellis, A. M. 2003, Chem. Phys. Lett., 374, 392 [Google Scholar]
- Wiesenfeld, L., & Faure, A. 2013, MNRAS, 432, 2573 [NASA ADS] [CrossRef] [Google Scholar]
- Wiesenfeld, L., Scribano, Y., & Faure, A. 2011, Phys. Chem. Chem. Phys., 13, 8230 [NASA ADS] [CrossRef] [Google Scholar]
- Zakharenko, O., Motiyenko, R. A., Margulès, L., & Huet, T. R. 2015, J. Mol. Spectrosc., 317, 41 [NASA ADS] [CrossRef] [Google Scholar]
All Tables
All Figures
|
Fig. 1 Coordinate system used for the H2CO+He and HDCO+He complexes. In each case, the molecule lies in the x–z plane, with its centre of mass fixed at the origin. |
| In the text | |
|
Fig. 2 Interaction energies as a function of R , at θ = 80◦ and φ = 0◦, for the HDCO+He complex computed using CCSD(T) and CCSD(T)-F12 methods with different basis sets. The estimated CBSL energies are also shown for comparison. |
| In the text | |
|
Fig. 3 First radial coefficients νlm(R) of the expansion PES for H2CO+He (solid line), HDCO+He (dashed lines), and D2CO+He (dotted line). |
| In the text | |
|
Fig. 4 Contour plots for the H2CO + He (top panels) and HDCO + He (bottom panels) complexes at selected configurations. Positive energies are shown in red contour lines, while negative energies are shown in blue lines in steps of 3 cm−1. |
| In the text | |
|
Fig. 5 Contour plots of the potential energy surface for the H2CO+He (top panel) and HDCO+He (bottom panel) complexes with R relaxed in the [2.5, 5.0] Å interval. The contour lines are in steps of 2 cm−1. |
| In the text | |
|
Fig. 6 Rotational de-excitation cross sections as a function of the collision energy for transitions from the 30,3 state. Top panel: para-H2CO+He (solid lines) and ortho-D2CO+He (dotted lines). Bottom panel: HDCO+He. |
| In the text | |
|
Fig. 7 Rotational de-excitation rate coefficients (solid lines) as a function of temperature for rotational transitions of para-H2CO (top panel) and ortho-H2CO (bottom panel). Data from Green (1991) (dash–dotted lines), and the mass-scaled data of Wiesenfeld & Faure (2013) (dashed lines) are also included. |
| In the text | |
|
Fig. 8 Rotational de-excitation rate coefficients from the initial state 30,3, for all the allowed rotational transitions of D2CO (dashed line) and HDCO (solid line) induced by collisions with He, plotted as a function of the temperature. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.